专家共识4――血栓性微血管病性溶血性贫血的诊断和处理指南
2012-02-12 11:30:00
血栓性微血管病性溶血性贫血的诊断和处理指南英国血液学会冯晴郑茵红节译郑小凡审校
血栓性血小板减少性紫癜(TTP)最早由Moschowitz在1924年描述,多年来经典的五联征一直作为其诊断特征。然而,其他几个综合征如溶血性尿毒症综合征(HUS)、子痫(eclampsia)、HELLP综合征(溶血、肝功能指标升高、血小板减少)也有类似临床特征。因而认为尽管这些疾病的病理生理特征有所不同,但在临床表现上有一定的相互重叠性。最近随着对新的vonWillebrand因子(VWF)-金属蛋白酶活性特征的认识,以及该因子在某些微血管性溶血疾病中缺乏或有抑制因子存在,使不同患者的发病机制更加容易明确,并可能更迅速地得到合适的治疗。然而,还存在相当多的问题,如缺乏合理设计的随机临床试验,临床资料未分类和管理。其中部分原因在于患者就诊较分散,首诊医师包括血液学、产科、神经科医师和感染性疾病医师。这个指南的目的在于制定出该疾病的不同临床分型、公认的诊断标准和可供选择的严格的治疗方案。众所周知,目前还缺乏很好的研究结果来支持制定一些标准的方案。血栓性血小板减少性紫癜(TTP)
TTP的发病罕见,发病率3.7/百万人。TTP的及时诊断和处理至关重要,早期阶段治疗的延误对疾病预后存在负面影响。
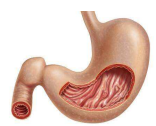
一、诊断和临床特征TTP是一种临床诊断。其特征为经典的五联征,即血小板减少、微血管性溶血性贫血(MAHA)、复杂多变的神经系统症状、肾脏损害和发热。常常起病凶险。神经系统症状表现多样,包括头痛、举止异常、一过性脑缺血发作(TIA)、癫痫和昏迷。昏迷的出现提示预后不良。其他症状还有胃肠缺血(表现为腹痛),浆液性视网膜剥离也认为与TTP有关。然而约有35%TTP患者不出现神经症状或体征。而HUS的三联征也包括急性肾功能不全、MAHA和血小板减少,因此会造成诊断不明确。另外,发热和肾功能损伤只存在于小部分患者中,因此临床上出现不明原因的MAHA和血小板减少时也可诊断为TTP。
二、临床分型TTP可以有许多不同的临床表现,临床亚型的不同可能会对疾病处理产生影响。先天性TTP又称之为SchulmanUpshaw综合征,罕见,通常发生于婴儿或少儿阶段。由位于vV/F-CP基因上的9q34染色体突变,使vWF-CP绝对缺乏所致。获得性TTP根据有无诱因,又可分为:①急性原发性TTP。多见,无明确病因,突然发病,病情发展迅速,约75%的患者在发病后3个月内死亡。②继发性TTP。有特殊的诱因可寻,如妊娠、恶性肿瘤、感染、HIV、骨髓移植后、自身免疫性疾病、药物(口服避孕药、噻氯匹定、环孢霉素、丝裂霉素)等。③间歇性(复发性)TTP。反复发作,复发间隔期不可预知,在1个月内复发者为近期复发,而在1个月后复发者为远期复发。
三、发病机制TTP发病机制中最主要的组织学异常是血小板微血管血栓的形成,且主要影响肾脏和脑血液循环,这决定了其临床表现。当先天性TTP患者的富血小板血浆(PRP)受到剪切压力时,就会发生过度的血小板凝集,这一过程由超大VWF多聚体(ULVWF)所介导,ULVWF并不是循环血浆中的正常成分,相反,ULVWF经蛋白水解后以较小的VWF聚合物形式参与循环。在正常血浆中,VWF除了主要的225kDa亚单位,还能检测到189,176和140kDa的片断。这是由于成熟亚基Tyr-842和Met-843残基间的单肽键劈裂所致。相同的片断可通过活体内存在的一种新的金属蛋白酶的作用下产生。最近已经证实这一蛋白酶属于ADAMTS家族(含Ⅰ型血小板结合蛋白模体的解聚蛋白和金属蛋白酶)中的新成员即ADAMTS13。这种VWF剪切蛋白酶(VWF-CP)活性的缺乏与获得性和先天性TTP有关。到目前为止,所有的原发性TTP都有严重的蛋白酶缺乏,而继发性TTP蛋白酶活性有可能正常。在一组111例的血栓性微血管病患者中,有66例被确诊为TTP(25例为原发性,41例为继发性),另外有45例诊断为HUS,TTP的蛋白酶缺乏试验敏感性为89%,特异性为91%。以往的报道认为先天性TTP是由于IgG亚型的抑制性自身抗体所致,但是在上述病例中,只有14例患者(56%)有蛋白酶抑制剂。而由于某种物质的缺乏所致的先天性TTP,可延迟到成人发病。同时,硬化病、尿毒症、急性感染、弥漫性血管内凝血(DIC)和恶性肿瘤也都有VWF-CP活性的降低。因此,尽管TTP对VWF-CP检测很敏感,但VWF-CP活性降低对TTP并无特异性。而且,这种模式不能解释血栓病的解剖学变化。内皮组织是一种异质性组织,受多种因子的调节,包括细胞因子、微环境)和剪切力。这些参数任何一种的变化都会影响VWF-CP本身活性或影响VWF对蛋白酶的敏感性。
本文部分内容来源于网络,如涉及版权问题请立即与微微健康网联系。